◼ Cyclic π-delocalization in metallacycle C7H7FeCl
This example comes from the original study
Electron delocalization in planar metallacycles: Hückel or Möbius aromatic?
D.W. Szczepanik (), M. Solà () ChemistryOpen 8 (2019) 219−227. DOI: 10.1002/open.201900014. URL
Abstract:
In this work the relationship between the formal number of π-electrons, d-orbital conjugation topology, π-electron delocalization and aromaticity in d-block metallacycles is investigated in the context of recent findings concerning the correlation of π-HOMO topology and the magnetic aromaticity indices in these species. It is demonstrated that for π-electron rich d-metallacycles the direct link between aromaticity, the number of π-electrons and the frontier π-orbital topology does not strictly hold and for such systems it is very difficult to unambiguously associate their aromaticity with the '4n+2' (Hückel) and '4n' (Möbius) rules. It is also shown that the recently proposed electron density of delocalized bonds (EDDB) method can successfully be used not only to quantify and visualize aromaticity in such difficult cases, but also – in contrast to magnetic aromaticity descriptors – to provide a great deal of information on the real role of d-orbitals in metallacycles without the ambiguity of bookkeeping of electrons in the π-subsystem of the molecular ring. Interestingly, some of the metallacycles studied cannot be classified exclusively as Hückel or Möbius because they have a hybrid Hückel-Möbius or even quasi-aromatic nature.
1. ORCA input files
To investigate bond delocalization effects in the C7H7FeCl metallacycle at the CAM-B3LYP/def2-SVP theory level, use the following input file:
! CAM-B3LYP def2-SVP
* xyz 0 1
Fe -0.064251938266 0.000000000000 0.383381287583
C 0.102879051008 1.467437846806 -0.581782848285
C 0.102879051008 -1.467437846806 -0.581782848285
C 0.042058436784 1.945591795572 -1.877580721536
C 0.042058436784 -1.945591795572 -1.877580721535
C -0.012197738912 1.304915633429 -3.124178523721
C -0.012197738912 -1.304915633430 -3.124178523721
C -0.022766917881 0.000000000000 -3.622055704846
H -0.061079076601 0.000000000000 -4.715595953142
H 0.167516111578 2.281514394810 0.169634195203
H 0.167516111578 -2.281514394810 0.169634195204
H 0.029959638034 3.038668532273 -1.974142731372
H 0.029959638034 -3.038668532273 -1.974142731372
H -0.055473924074 2.032073158396 -3.942519373400
H -0.055473924074 -2.032073158396 -3.942519373400
Cl 1.502523783826 0.000000000000 1.822684376427
*
Run the calculation using ORCA (tested version: 6.0.1) and convert the resulting c7h7fecl.gbw file to JSON using orca_2json to get c7h7fecl.json.
First, we are going to investigate the distribution of electron density of all delocalized bonds in the entire molecule using the EDDBG function (the subscript G stands for global: all atoms and bonds in the molecule are included). To do so, run the following command in the terminal:
terminal
$ runEDDB --input c7h7fecl.json --quiet
The --quiet (or -q) option enables quiet mode: only the EDDBG summary populations are printed; useful for batch jobs where only the numerical result is needed:
output
> Printing summary of EDDB calculations:
Total ED_G population: 49.79373 per atom: 3.11211
Total EDLB_G population: 41.50545 per atom: 2.59409
Total EDDB_G population: 8.28827 per atom: 0.51802
> Calculation completed on 2026-05-11. Total time: 0s.
The default analysis basis is the Natural Valence Basis (NVB), which retains only the valence-shell Natural Atomic Orbitals (NAO). Of the ~49.8 valence electrons in the NVB, about 41.5e are localized (lone pairs and two-center bonds) and ~8.3e are shared through the system of delocalized bonds.
To quickly visualize the EDDBG-layer of the one-electron density, add the --output-eddb option:
This generates a Gaussian cube file (c7h7fecl.EDDB.cube) for visualization in, e.g., Avogadro2, IQmol, GaussView, VMD, or Chemcraft; a typical isosurface value is 0.015-0.020. The --ncores (or -n) option parallelizes the grid evaluation, which is the most compute-intensive step. It is recommended to use as many cores as available.
The grid quality is controlled by --cube-size (or -c) option (default: 100). Running without --quiet shows the cube export details:
output
> Exporting EDDB_G to c7h7fecl.EDDB.cube...
Grid scope: Global
Grid size: 74x115x120 (~100^3)
Progress: ######################################## 100%
Integrated density: 8.28712 (100.0% of 8.28827)
Time: 3s
The integrated density (~8.287e) almost perfectly matches the analytical EDDBG population (~8.288e) — this confirms the cube accurately represents the density.
To visualize the generated cube file (c7h7fecl.EDDB.cube) in Avogadro2, select in program menu Analyze → Create Surfaces and set parameters as in the figure below:
3. Detailed population analysis
Now, switch to the verbose mode using --verbose (or -v) option:
terminal
$ runEDDB --input c7h7fecl.json --verbose
First, the program reports what was parsed from the wavefunction file:
output
> Loading and parsing input file... 0s
Source file: c7h7fecl.json
File format: ORCA JSON
Number of atoms: 16
Number of basis functions: 182 (spherical)
Number of contracted shells: 82
Number of primitives: 166
Number of molecular orbitals: 182
Effective core potential: No
Charge / multiplicity: 0 / 1
Total electrons: 92
Unrestricted: No
Reference type: HF / DFT
Electron population (Tr[D*S]): 92.00000
Next, it summarises the internal AO→NAO localization procedure:
output
> Building natural atomic orbitals (NAO)... 0s
Total number of NAOs: 182 = 21 (Cor) + 45 (Val) + 116 (Ryd)
Total electron population: 92.00000
- core population: 41.99888 (~45.7 %)
- valence population: 49.79373 (~54.1 %)
- rydberg population: 0.20739 (~0.2 %)
Target NAO representation: Natural valence basis (NVB = Val)
- NVB population: 49.79373 (~54.1 %)
Out of the 182 NAOs constructed by the internal driver, 21 are classified as core (Cor), 45 as valence (Val), and 116 as Rydberg (Ryd). The default NAO representation (NVB) keeps only the 45 valence NAOs that capture ~49.8e — the analysis below operates on this subspace.
In the next stage program prints the BOP configuration and performs calculations:
output
> Performing multicenter bond-orbital projections (BOP)...
Type of EDDB function: EDDB_G (entire molecular system)
BOP algorithm: Standard BOP (HF/DFT 1-densities)
Atoms included in BOP: 16
Pairs included in BOP: 69
Bond-order threshold: 0.00100
Progress: ######################################## 100%
Number of BOP cycles: 1
BOP time: 0s
In the entire system, 69 atom pairs exceed the Wiberg bond-order threshold (10−3) and enter the BOP cycle.
After the BOP step, program prints three electron-population tables. The first lists Natural Orbitals for Bond Delocalization (NOBD):
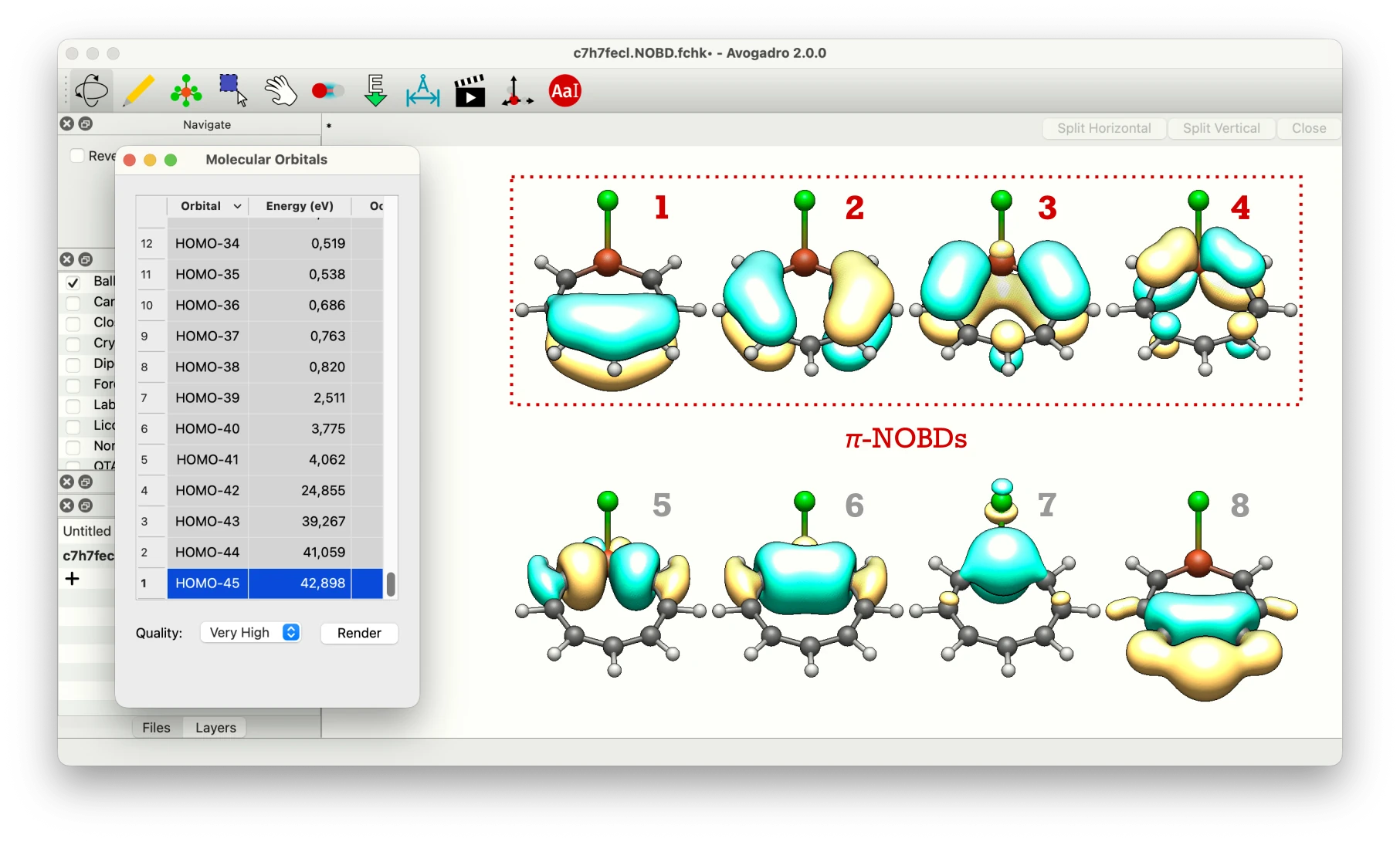
From the table above it is clear that the first four NOBDs make the dominant contributions to electron delocalization. Their orbital decomposition shows that the first three NOBDs have almost purely π-symmetry (~1.7-1.8e, 98-100% p-character), while the fourth involves the Fe atom through its 3d orbitals (~26% p- and ~74% d-character). The remaining NOBDs have much smaller occupancies associated with σ-delocalization tails.
The next table presents the population analysis in the atomic-orbital resolution — one row per NAO in the target representation (here NVB, valence-only):
output
> Printing results of electron population analysis in orbital resolution...
+------------+-------------+---------------------------+-----------------------------+
| Atom | Orbital | Electron delocalization | Electron population |
+------------+-------------+---------------------------+-----------------------------+
Index Sym Index NVB Total Alpha Beta Total Alpha Beta
----- --- ----- ---- ======= ------- ------- ------- ------- -------
1 Fe 1 4s 0.04480 - - 0.41469 - -
1 Fe 2 3d1 0.05243 - - 1.92913 - -
1 Fe 3 3d2 0.08352 - - 1.14207 - -
1 Fe 4 3d3 0.14951 - - 0.93536 - -
1 Fe 5 3d4 0.12314 - - 1.33084 - -
1 Fe 6 3d5 0.88862 - - 1.46618 - -
2 C 7 2s 0.06676 - - 1.15495 - -
2 C 8 2p1 0.64327 - - 0.83275 - -
2 C 9 2p2 0.13813 - - 1.20113 - -
2 C 10 2p3 0.03810 - - 1.10601 - -
3 C 11 2s 0.06676 - - 1.15495 - -
3 C 12 2p1 0.64327 - - 0.83275 - -
3 C 13 2p2 0.13813 - - 1.20113 - -
3 C 14 2p3 0.03810 - - 1.10601 - -
4 C 15 2s 0.03170 - - 0.96152 - -
4 C 16 2p1 0.86467 - - 1.01977 - -
4 C 17 2p2 0.04565 - - 1.19031 - -
4 C 18 2p3 0.04158 - - 1.10315 - -
5 C 19 2s 0.03170 - - 0.96152 - -
5 C 20 2p1 0.86467 - - 1.01977 - -
5 C 21 2p2 0.04565 - - 1.19031 - -
5 C 22 2p3 0.04158 - - 1.10315 - -
6 C 23 2s 0.03189 - - 0.94729 - -
6 C 24 2p1 0.79166 - - 0.89829 - -
6 C 25 2p2 0.03619 - - 1.12827 - -
6 C 26 2p3 0.03839 - - 1.14204 - -
7 C 27 2s 0.03189 - - 0.94729 - -
7 C 28 2p1 0.79166 - - 0.89829 - -
7 C 29 2p2 0.03619 - - 1.12827 - -
7 C 30 2p3 0.03839 - - 1.14204 - -
8 C 31 2s 0.03027 - - 0.93831 - -
8 C 32 2p1 0.93201 - - 1.02360 - -
8 C 33 2p2 0.03868 - - 1.08621 - -
8 C 34 2p3 0.03699 - - 1.18657 - -
9 H 35 1s 0.02106 - - 0.77162 - -
10 H 36 1s 0.02297 - - 0.79925 - -
11 H 37 1s 0.02297 - - 0.79925 - -
12 H 38 1s 0.02647 - - 0.77648 - -
13 H 39 1s 0.02647 - - 0.77648 - -
14 H 40 1s 0.01933 - - 0.77340 - -
15 H 41 1s 0.01933 - - 0.77340 - -
16 Cl 42 3s 0.01559 - - 1.95190 - -
16 Cl 43 3p1 0.03425 - - 1.85294 - -
16 Cl 44 3p2 0.09281 - - 1.93720 - -
16 Cl 45 3p3 0.07112 - - 1.75792 - -
----- --- ----- ---- ======= ------- ------- ------- ------- -------
The data reveal that the main delocalization channel involves the 3d5 orbital on Fe, occupied by ~1.5e and contributing ~0.9e (~60%) to delocalization, together with the 2p1 orbitals on carbon atoms, occupied by about 0.8–1.0e and contributing about 0.6–0.9e to delocalization. Electrons on chlorine and hydrogen atoms are predominantly localized.
Finally, the last table collapses the atomic-orbital data to atomic resolution:
output
> Printing results of electron population analysis in atomic resolution...
+------------+-----------------------------------+-----------------------------------+
| Atom | Electron delocalization | Electron population |
+------------+-----------------------------------+-----------------------------------+
Index Sym Total Alpha Beta Total Alpha Beta
----- --- ========= --------- --------- --------- --------- ---------
1 Fe 1.34200 - - 7.21828 - -
2 C 0.88626 - - 4.29484 - -
3 C 0.88626 - - 4.29484 - -
4 C 0.98361 - - 4.27474 - -
5 C 0.98361 - - 4.27474 - -
6 C 0.89812 - - 4.11588 - -
7 C 0.89812 - - 4.11588 - -
8 C 1.03795 - - 4.23468 - -
9 H 0.02106 - - 0.77162 - -
10 H 0.02297 - - 0.79925 - -
11 H 0.02297 - - 0.79925 - -
12 H 0.02647 - - 0.77648 - -
13 H 0.02647 - - 0.77648 - -
14 H 0.01933 - - 0.77340 - -
15 H 0.01933 - - 0.77340 - -
16 Cl 0.21377 - - 7.49995 - -
----- --- ========= --------- --------- --------- --------- ---------
The numbers confirm that only Fe and the C atoms in the ring contribute significantly to the global delocalization. This atomic-resolution table is the only one printed by default (i.e., without --verbose option); it is the starting point for most discussions and is sufficient for many quick assessments.
4. Cyclic delocalization of π-electrons
Aromatic stabilization in molecular rings with π-conjugated bonds is associated with cyclic delocalization of π-electrons, whereas cross-ring orbital interactions are often locally destabilizing (cf. the antibonding interaction between para-related carbon atoms in benzene). To assess the effect of cyclic π-electron delocalization in the 8-membered ring (8MR) of the C7H7FeCl system, we perform an EDDBP calculation (the subscript P stands for pathway):
The --pathway (or -p) option specifies the bond connectivity along the ring. The --output-nobd option generates a new Gaussian FCHK file (c7h7fecl.NOBD.fchk) in which the Alpha MO coefficients are replaced by the spinless NOBDs. The --auto-pi (or -a) option automatically selects only π-type NOBDs based on their orbital composition; also, this flag triggers printing of the NOBD-resolution table by default, so one can immediately see which orbitals were classified as π-type.
The --auto-pi option correctly identifies the first four NOBDs as π-orbitals (marked with *); together they account for ~8.0e π-electrons of which ~5.4e is cyclically delocalized. To visually inspect the NOBDs and verify the automatic π-NOBD selection, open the generated c7h7fecl.NOBD.fchk file in Avogadro2 and select particular NOBD in Molecular Orbitals window (in generated c7h7fecl.NOBD.fchk file α-MOs ordered by increasing energy are replaced by NOBDs ordered by decreasing occupation number):
As follows from the summary of EDDB calculations, the average per-atom population of cyclically delocalized π-electrons in the 8MR is 0.68002e. However, the EDDBP atomic populations reveal that the distribution of electrons is not uniform: a noticeable π-deficiency on atoms 2 and 3 is a fingerprint of high-energy ionic resonance structures contributing to the wavefunction and reducing the aromatic stabilization energy. In this sense, the capacity of the π-channel to connect all eight atomic orbitals in a coherent cycle is limited by those two carbon atoms.
The Cyclic Delocalization Index (CDI), printed at the end of the summary, equals the smallest per-atom EDDBP value — the maximum population of π-electrons (per atom) that can be uniformly delocalized around the cycle. Here, the cyclic delocalization index is about ~0.54; for comparison, the CDI for benzene is ~0.89, so the relative π-aromaticity in the 8MR of C7H7FeCl can be estimated as ~61%.
5. 3d-orbital conjugation topology
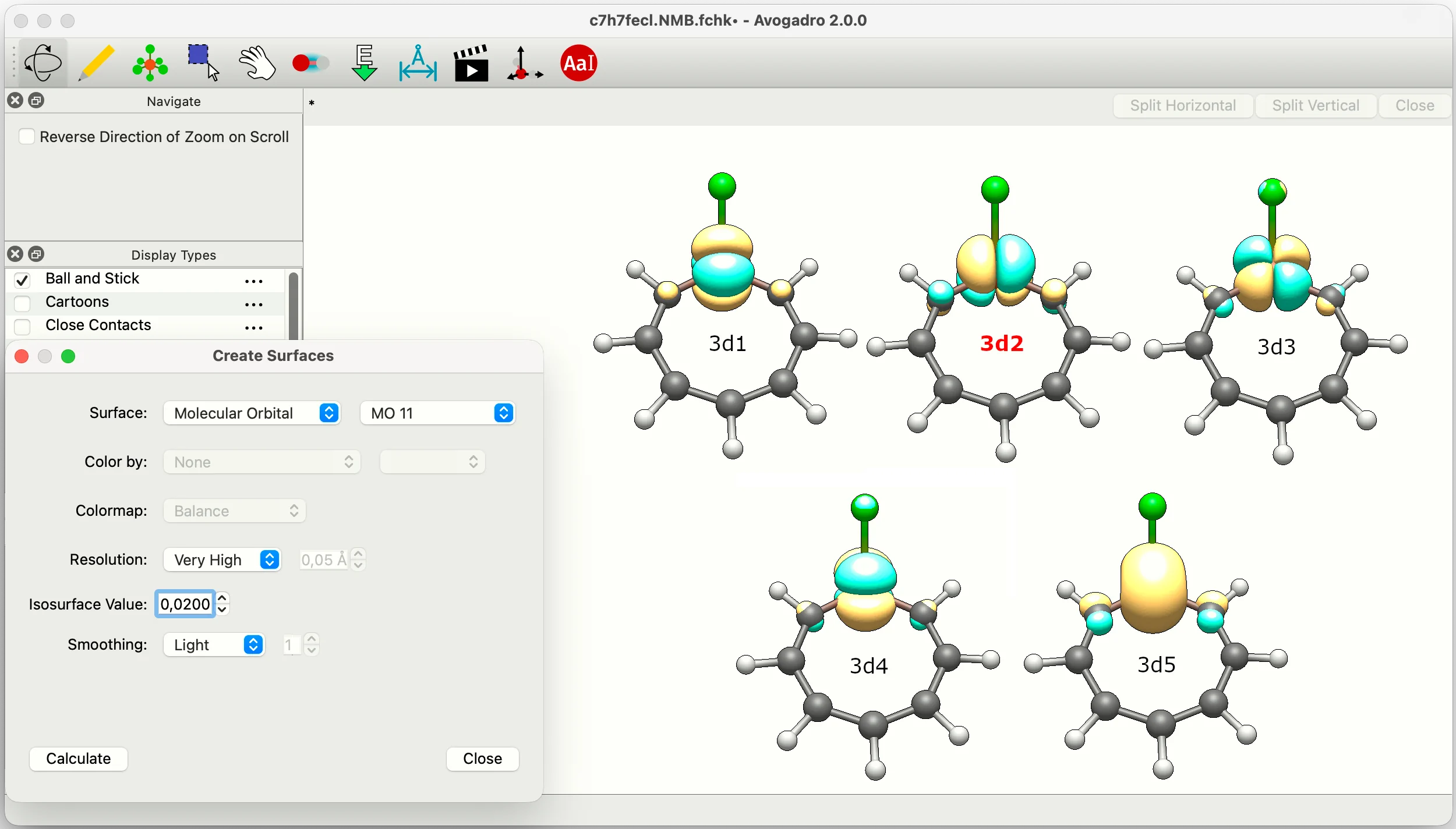
The remaining open question is: what does the 3d5 orbital on Fe look like? To find out, simply run:
and open the generated c7h7fecl.NVB.fchk file in Avogadro2 (select Analyze → Create Surfaces). According to the resulting NVB-resolution table,
output
> Printing results of electron population analysis in orbital resolution...
+------------+-------------+---------------------------+-----------------------------+
| Atom | Orbital | Electron delocalization | Electron population |
+------------+-------------+---------------------------+-----------------------------+
Index Sym Index NVB Total Alpha Beta Total Alpha Beta
----- --- ----- ---- ======= ------- ------- ------- ------- -------
1 Fe 1 4s 0.00827 - - 0.01135 - -
1 Fe 2 3d1 0.00040 - - 0.00055 - -
1 Fe 3 3d2 0.00929 - - 0.01275 - -
1 Fe 4 3d3 0.02218 - - 0.04828 - -
1 Fe 5 3d4 0.00517 - - 0.00709 - -
1 Fe 6 3d5 0.65406 - - 1.42504 - -
2 C 7 2s 0.00029 - - 0.00052 - -
2 C 8 2p1 0.54108 - - 0.81631 - -
2 C 9 2p2 0.00004 - - 0.00008 - -
2 C 10 2p3 0.00106 - - 0.00164 - -
3 C 11 2s 0.00029 - - 0.00052 - -
3 C 12 2p1 0.54108 - - 0.81631 - -
3 C 13 2p2 0.00004 - - 0.00008 - -
3 C 14 2p3 0.00106 - - 0.00164 - -
4 C 15 2s 0.00001 - - 0.00001 - -
4 C 16 2p1 0.74829 - - 1.00758 - -
4 C 17 2p2 0.00002 - - 0.00003 - -
4 C 18 2p3 0.00239 - - 0.00322 - -
5 C 19 2s 0.00001 - - 0.00001 - -
5 C 20 2p1 0.74829 - - 1.00758 - -
5 C 21 2p2 0.00002 - - 0.00003 - -
5 C 22 2p3 0.00239 - - 0.00322 - -
6 C 23 2s 0.00000 - - 0.00000 - -
6 C 24 2p1 0.67629 - - 0.89445 - -
6 C 25 2p2 0.00008 - - 0.00011 - -
6 C 26 2p3 0.00115 - - 0.00152 - -
7 C 27 2s 0.00000 - - 0.00000 - -
7 C 28 2p1 0.67629 - - 0.89445 - -
7 C 29 2p2 0.00008 - - 0.00011 - -
7 C 30 2p3 0.00115 - - 0.00152 - -
8 C 31 2s 0.00000 - - 0.00000 - -
8 C 32 2p1 0.79833 - - 1.01672 - -
8 C 33 2p2 0.00000 - - 0.00000 - -
8 C 34 2p3 0.00101 - - 0.00129 - -
----- --- ----- ---- ======= ------- ------- ------- ------- -------
the 3d5 (Fe) orbital has index 6. A visual inspection of Molecular Orbital no. 6 reveals this is a 3dXY orbital, which means the metallacycle is a Möbius-Craig aromatic with a predominantly δ-type conjugation topology (see the reference paper for more examples of metallacycles with π- and δ-conjugation topology).
6. Final conclusion
The EDDB analysis at the CAM-B3LYP/def2-SVP theory level reveals that the model C7H7FeCl metallacycle (in its ground-state configuration) can be classified as an 8π-electron Möbius-Craig aromatic species with predominating δ-conjugation topology involving 3dXY (Fe) orbital. The effectiveness of cyclic delocalization of π-bonds in this 8-membered ring is relatively high - about 61% of the one in archetypical π-aromatic benzene.