
aromaticity.uj.edu.pl
Szczepanik Research Group
Department of Theoretical Chemistry
Faculty of Chemistry, Jagiellonian University
Gronostajowa 2, 30-387 Krakow, Poland
Tel: (+48) 12 686 23 90
E-mail: dariusz.szczepanik@uj.edu.pl![]()
ABOUT US PROJECTS PUBLICATIONS EDDB TUTORIALS X
About us People Featured papers Featured posters
◼ About us
We develop modern theoretical approaches to assess chemical bonding structure and reactivity of π-conjugated systems, including:
- Ground-state projected covalency index (GSPCI); URL
- Correlation analysis of chemical bonds (CACB); URL
- Bond-orbital projection method (BOP); URL
- Electron density of delocalized bonds (EDDB); URL
- Bond delocalization function (BDF). URL
- Asymmetric orthogonalization schemes in the Hilbert space; URL
- Markov's stationarity of the second-order density matrices; URL
- Shannon's algebra of atomic-orbital communication channels; URL
- Information-entropy representation of electron correlation; URL
- Hybrid extrapolation scemes for robust CCSD(T)/CBS estimates. URL
◼ People

|
Dr hab. Dariusz W. Szczepanik  Principal investigator Introducing Dariusz W. Szczepanik Position: Adjunct with habilitation Affiliation: Research Group of Thermodynamics and Dynamics of Chemical Reactions, Kazimierz Guminski Department of Theoretical Chemistry, Faculty of Chemistry, Jagiellonian University |
Senior researcher: ◼ Dr hab. Marcin Andrzejak Postdoc: ◼ Dr Ouissam El Bakouri Students: ◼ MSc Mikolaj Dratwinski ◼ MSc Pawel A. Wieczorkiewicz ◼ MSc Joanna Zams ◼ BSc Weronika Sitkowska |
◼ Featured papers
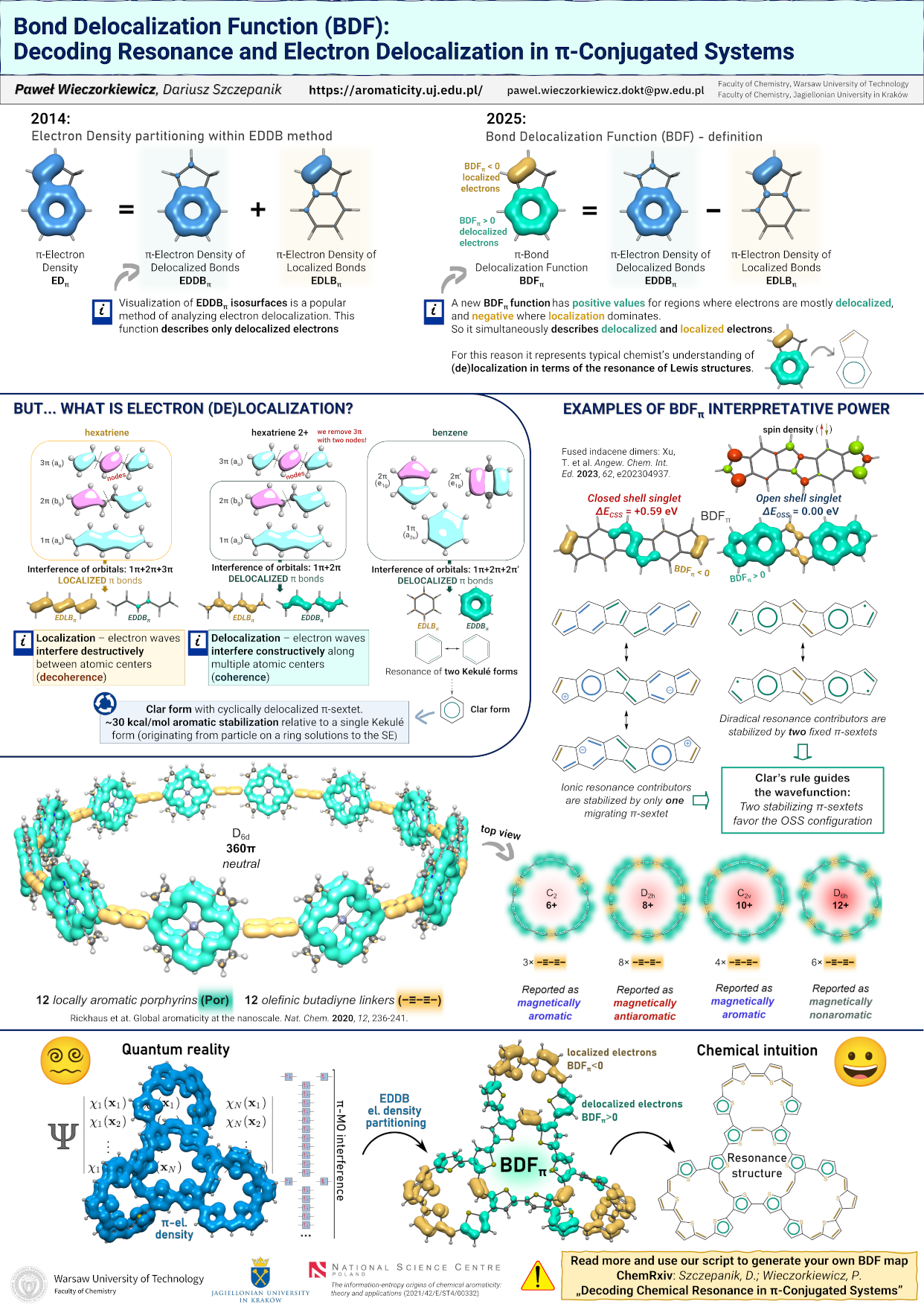
Decoding chemical resonance in π-conjugated systems
D.W. Szczepanik (), P. Wieczorkiewicz ()
ChemRXiv (2025) DOI: 10.26434/chemrxiv-2025-m80qm-v3. URL
Abstract:
Understanding chemical resonance in poly- and macrocyclic π-conjugated systems requires access to the full topology of ground-state electron delocalization — not merely its observable molecular response. While magnetically-induced ring currents emerge from a narrow subset of frontier orbitals, as dictated by the Fowler–Steiner selection rules, the magnetically silent electrons that define the deeper resonance structure remain hidden from view. Here, we present a wavefunction-based framework that maps the spatial distribution and coherence of π-electron density across bonding domains, revealing patterns of delocalization that transcend the current-based indicators. Applied to a range of π-systems widely discussed in the context of global (anti)aromaticity, this approach exposes the internal logic of chemical resonance — distinguishing topological coherence from superficial circulation. Our findings suggest a shift in perspective: aromaticity arises not from current, but from coherence — the quiet entanglement of electrons bound by phase, not motion.
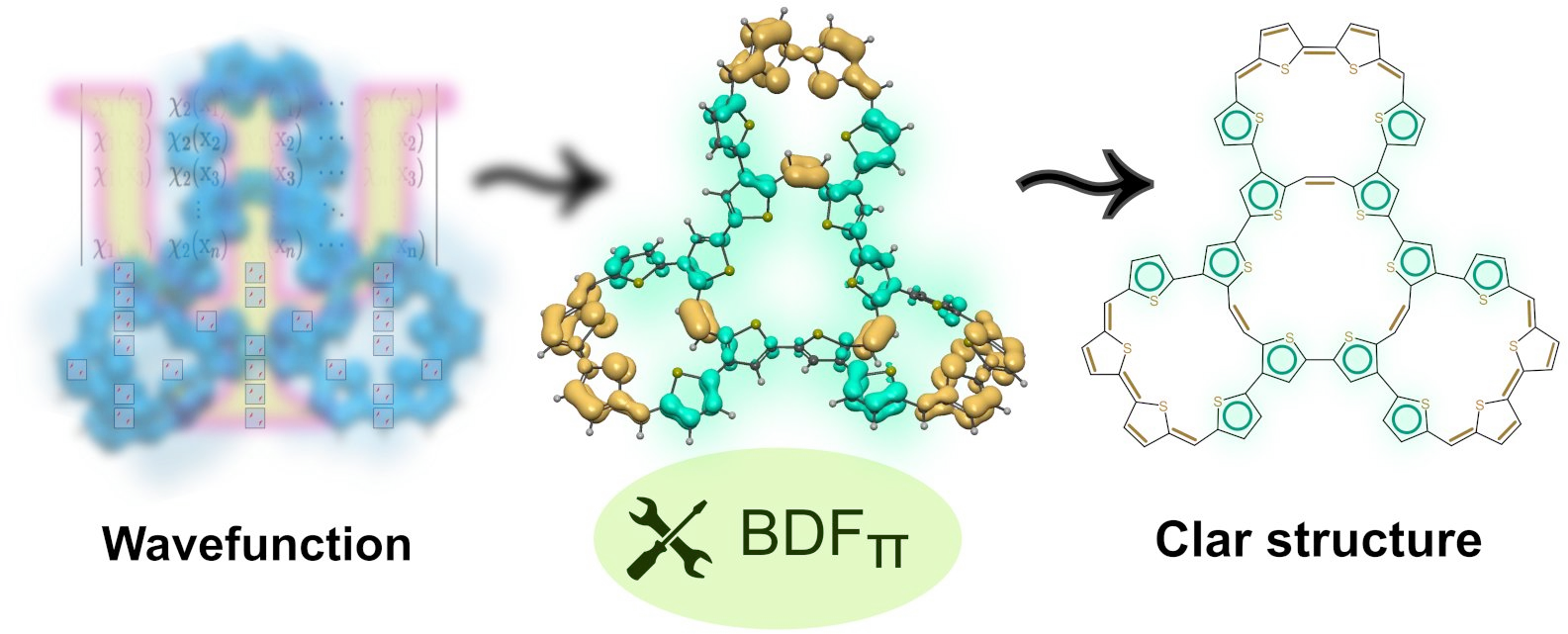
D.W. Szczepanik (), P. Wieczorkiewicz ()
ChemRXiv (2025) DOI: 10.26434/chemrxiv-2025-m80qm-v3. URL
Abstract:
Understanding chemical resonance in poly- and macrocyclic π-conjugated systems requires access to the full topology of ground-state electron delocalization — not merely its observable molecular response. While magnetically-induced ring currents emerge from a narrow subset of frontier orbitals, as dictated by the Fowler–Steiner selection rules, the magnetically silent electrons that define the deeper resonance structure remain hidden from view. Here, we present a wavefunction-based framework that maps the spatial distribution and coherence of π-electron density across bonding domains, revealing patterns of delocalization that transcend the current-based indicators. Applied to a range of π-systems widely discussed in the context of global (anti)aromaticity, this approach exposes the internal logic of chemical resonance — distinguishing topological coherence from superficial circulation. Our findings suggest a shift in perspective: aromaticity arises not from current, but from coherence — the quiet entanglement of electrons bound by phase, not motion.
Three-dimensional fully π-conjugated macrocycles: when 3D-aromatic and when 2D-aromatic-in-3D?
O. El Bakouri, D.W. Szczepanik, K. Jorner, R. Ayub, P. Bultinck, M. Solà (), H. Ottosson ()
Journal of the Americal Chemical Society 144 (2022) 8560−8575. DOI: 10.1021/jacs.1c13478. URL
Abstract:
Several fully π-conjugated macrocycles with puckered or cage-type structures were recently found to exhibit aromatic character according to both experiments and computations. We examine their electronic structures and put them in relation to 3D-aromatic molecules (e.g., closo-boranes) and to 2D-aromatic polycyclic aromatic hydrocarbons. Using qualitative theory combined with quantum chemical calculations, we find that the macrocycles explored hitherto should be described as 2D-aromatic with three-dimensional molecular structures (abbr. 2D-aromatic-in-3D) and not as truly 3D-aromatic. 3D-aromatic molecules have highly symmetric structures (or nearly so), leading to (at least) triply degenerate molecular orbitals, and for tetrahedral or octahedral molecules, an aromatic closed-shell electronic structure with 6n + 2 electrons. Conversely, 2D-aromatic-in-3D structures exhibit aromaticity that results from the fulfillment of Hückel's 4n + 2 rule for each macrocyclic path, yet their π-electron counts are coincidentally 6n + 2 numbers for macrocycles with three tethers of equal lengths. It is notable that 2D-aromatic-in-3D macrocyclic cages can be aromatic with tethers of different lengths, i.e., with π-electron counts different from 6n + 2, and they are related to naphthalene. Finally, we identify tetrahedral and cubic π-conjugated molecules that fulfill the 6n + 2 rule and exhibit significant electron delocalization. Yet, their properties resemble those of analogous compounds with electron counts that differ from 6n + 2. Thus, despite the fact that these molecules show substantial π-electron delocalization, they cannot be classified as true 3D-aromatics.
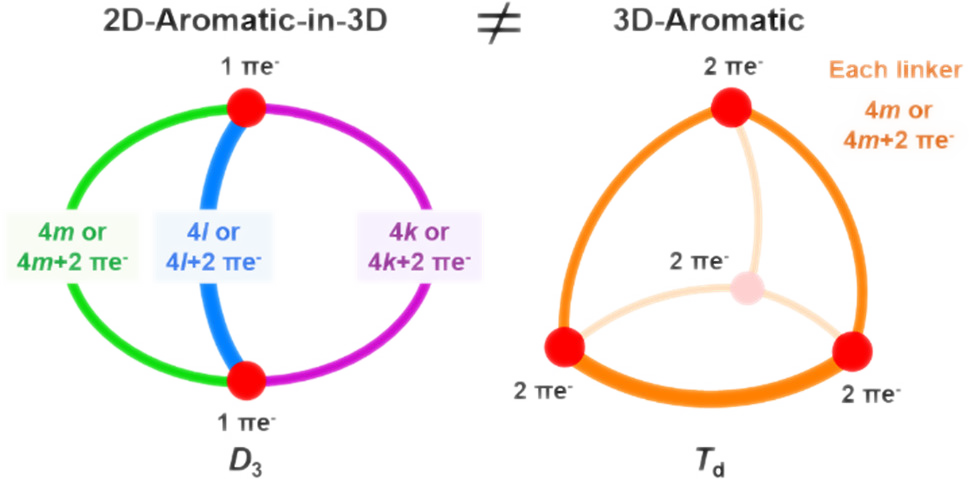
O. El Bakouri, D.W. Szczepanik, K. Jorner, R. Ayub, P. Bultinck, M. Solà (), H. Ottosson ()
Journal of the Americal Chemical Society 144 (2022) 8560−8575. DOI: 10.1021/jacs.1c13478. URL
Abstract:
Several fully π-conjugated macrocycles with puckered or cage-type structures were recently found to exhibit aromatic character according to both experiments and computations. We examine their electronic structures and put them in relation to 3D-aromatic molecules (e.g., closo-boranes) and to 2D-aromatic polycyclic aromatic hydrocarbons. Using qualitative theory combined with quantum chemical calculations, we find that the macrocycles explored hitherto should be described as 2D-aromatic with three-dimensional molecular structures (abbr. 2D-aromatic-in-3D) and not as truly 3D-aromatic. 3D-aromatic molecules have highly symmetric structures (or nearly so), leading to (at least) triply degenerate molecular orbitals, and for tetrahedral or octahedral molecules, an aromatic closed-shell electronic structure with 6n + 2 electrons. Conversely, 2D-aromatic-in-3D structures exhibit aromaticity that results from the fulfillment of Hückel's 4n + 2 rule for each macrocyclic path, yet their π-electron counts are coincidentally 6n + 2 numbers for macrocycles with three tethers of equal lengths. It is notable that 2D-aromatic-in-3D macrocyclic cages can be aromatic with tethers of different lengths, i.e., with π-electron counts different from 6n + 2, and they are related to naphthalene. Finally, we identify tetrahedral and cubic π-conjugated molecules that fulfill the 6n + 2 rule and exhibit significant electron delocalization. Yet, their properties resemble those of analogous compounds with electron counts that differ from 6n + 2. Thus, despite the fact that these molecules show substantial π-electron delocalization, they cannot be classified as true 3D-aromatics.
Excited state character of Cibalackrot-type compounds interpreted in terms of Hückel-aromaticity: a rationale for singlet fission chromophore design
W. Zeng, O. El Bakouri, D.W. Szczepanik (), H. Bronstein (), H. Ottosson ()
Chemical Science 12 (2021) 6159−6171. DOI: 10.1039/D1SC00382H. URL
Abstract:
The exact energies of the lowest singlet and triplet excited states in organic chromophores are crucial to their performance in optoelectronic devices. The possibility of utilizing singlet fission to enhance the performance of photovoltaic devices has resulted in a wide demand for tuneable, stable organic chromophores with wide S1-T1 energy gaps (>1 eV). Cibalackrot-type compounds were recently considered to have favorably positioned excited state energies for singlet fission, and they were found to have a degree of aromaticity in the lowest triplet excited state (T1). This work reports on a revised and deepened theoretical analysis taking into account the excited state Hückel-aromatic (instead of Baird-aromatic) as well as diradical characters, with the aim to design new organic chromophores based on this scaffold in a rational way starting from qualitative theory. We demonstrate that the substituent strategy can effectively adjust the spin distribution on the chromophore and thereby manipulate the excited state energy levels. Additionally, the improved understanding of the aromatic characters enables us to demonstrate a feasible design strategy to vary the excited state energy levels by tuning the number and nature of Hückel-aromatic units in the excited state. Finally, our study elucidates the complications and pitfalls of the excited state aromaticity and antiaromaticity concepts, highlighting that quantitative results from quantum chemical calculations of various aromaticity indices must be linked with qualitative theoretical analysis of the character of the excited states.
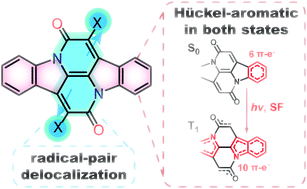
W. Zeng, O. El Bakouri, D.W. Szczepanik (), H. Bronstein (), H. Ottosson ()
Chemical Science 12 (2021) 6159−6171. DOI: 10.1039/D1SC00382H. URL
Abstract:
The exact energies of the lowest singlet and triplet excited states in organic chromophores are crucial to their performance in optoelectronic devices. The possibility of utilizing singlet fission to enhance the performance of photovoltaic devices has resulted in a wide demand for tuneable, stable organic chromophores with wide S1-T1 energy gaps (>1 eV). Cibalackrot-type compounds were recently considered to have favorably positioned excited state energies for singlet fission, and they were found to have a degree of aromaticity in the lowest triplet excited state (T1). This work reports on a revised and deepened theoretical analysis taking into account the excited state Hückel-aromatic (instead of Baird-aromatic) as well as diradical characters, with the aim to design new organic chromophores based on this scaffold in a rational way starting from qualitative theory. We demonstrate that the substituent strategy can effectively adjust the spin distribution on the chromophore and thereby manipulate the excited state energy levels. Additionally, the improved understanding of the aromatic characters enables us to demonstrate a feasible design strategy to vary the excited state energy levels by tuning the number and nature of Hückel-aromatic units in the excited state. Finally, our study elucidates the complications and pitfalls of the excited state aromaticity and antiaromaticity concepts, highlighting that quantitative results from quantum chemical calculations of various aromaticity indices must be linked with qualitative theoretical analysis of the character of the excited states.
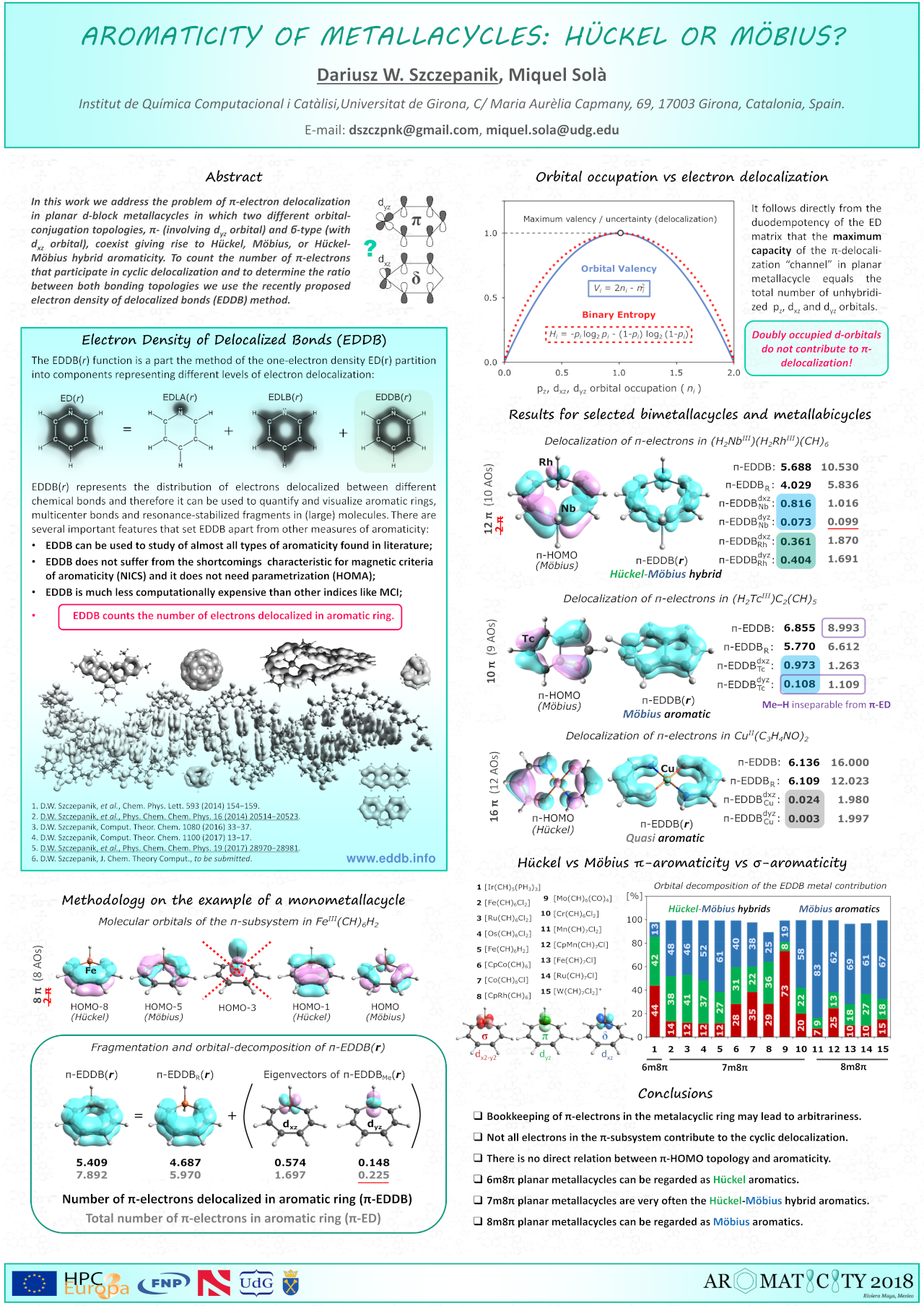
Electron delocalization in planar metallacycles: Hückel or Möbius aromatic?
D.W. Szczepanik (), M. Solà ()
ChemistryOpen 8 (2019) 219−227. DOI: 10.1002/open.201900014. URL
Abstract:
In this work the relationship between the formal number of π-electrons, d-orbital conjugation topology, π-electron delocalization and aromaticity in d-block metallacycles is investigated in the context of recent findings concerning the correlation of π-HOMO topology and the magnetic aromaticity indices in these species. It is demonstrated that for π-electron rich d-metallacycles the direct link between aromaticity, the number of π-electrons and the frontier π-orbital topology does not strictly hold and for such systems it is very difficult to unambiguously associate their aromaticity with the '4n+2' (Hückel) and '4n' (Möbius) rules. It is also shown that the recently proposed electron density of delocalized bonds (EDDB) method can successfully be used not only to quantify and visualize aromaticity in such difficult cases, but also – in contrast to magnetic aromaticity descriptors – to provide a great deal of information on the real role of d-orbitals in metallacycles without the ambiguity of bookkeeping of electrons in the π-subsystem of the molecular ring. Interestingly, some of the metallacycles studied cannot be classified exclusively as Hückel or Möbius because they have a hybrid Hückel-Möbius or even quasi-aromatic nature.
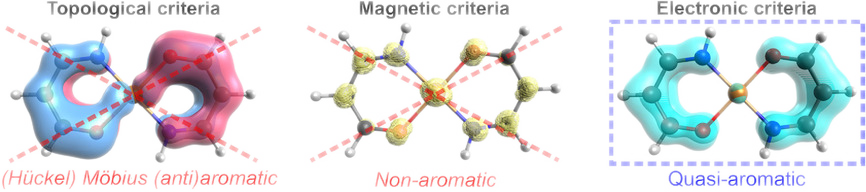
D.W. Szczepanik (), M. Solà ()
ChemistryOpen 8 (2019) 219−227. DOI: 10.1002/open.201900014. URL
Abstract:
In this work the relationship between the formal number of π-electrons, d-orbital conjugation topology, π-electron delocalization and aromaticity in d-block metallacycles is investigated in the context of recent findings concerning the correlation of π-HOMO topology and the magnetic aromaticity indices in these species. It is demonstrated that for π-electron rich d-metallacycles the direct link between aromaticity, the number of π-electrons and the frontier π-orbital topology does not strictly hold and for such systems it is very difficult to unambiguously associate their aromaticity with the '4n+2' (Hückel) and '4n' (Möbius) rules. It is also shown that the recently proposed electron density of delocalized bonds (EDDB) method can successfully be used not only to quantify and visualize aromaticity in such difficult cases, but also – in contrast to magnetic aromaticity descriptors – to provide a great deal of information on the real role of d-orbitals in metallacycles without the ambiguity of bookkeeping of electrons in the π-subsystem of the molecular ring. Interestingly, some of the metallacycles studied cannot be classified exclusively as Hückel or Möbius because they have a hybrid Hückel-Möbius or even quasi-aromatic nature.
The electron density of delocalized bonds (EDDB) applied for quantifying aromaticity
D.W. Szczepanik (), M. Andrzejak, J. Dominikowska, B. Pawelek, T.M. Krygowski, H. Szatylowicz, M. Solà
Physical Chemistry Chemical Physics 19 (2017) 28970−28981. DOI: 10.1039/C7CP06114E. URL
Abstract:
In this study the recently developed electron density of delocalized bonds (EDDB) is used to define a new measure of aromaticity in molecular rings. The relationships between bond-length alternation, electron delocalization and diatropicity of the induced ring current are investigated for a test set of representative molecular rings by means of correlation and principal component analyses involving the most popular aromaticity descriptors based on structural, electronic, and magnetic criteria. Additionally, a qualitative comparison is made between EDDB and the magnetically induced ring-current density maps from the ipsocentric approach for a series of linear acenes. Special emphasis is given to the comparative study of the description of cyclic delocalization of electrons in a wide range of organic aromatics in terms of the kekulean multicenter index KMCI and the newly proposed EDDBk index.
D.W. Szczepanik (), M. Andrzejak, J. Dominikowska, B. Pawelek, T.M. Krygowski, H. Szatylowicz, M. Solà
Physical Chemistry Chemical Physics 19 (2017) 28970−28981. DOI: 10.1039/C7CP06114E. URL
Abstract:
In this study the recently developed electron density of delocalized bonds (EDDB) is used to define a new measure of aromaticity in molecular rings. The relationships between bond-length alternation, electron delocalization and diatropicity of the induced ring current are investigated for a test set of representative molecular rings by means of correlation and principal component analyses involving the most popular aromaticity descriptors based on structural, electronic, and magnetic criteria. Additionally, a qualitative comparison is made between EDDB and the magnetically induced ring-current density maps from the ipsocentric approach for a series of linear acenes. Special emphasis is given to the comparative study of the description of cyclic delocalization of electrons in a wide range of organic aromatics in terms of the kekulean multicenter index KMCI and the newly proposed EDDBk index.
◼ Featured posters
ESOR2025 - European Symposium on Organic Reactivity (Padova, Italy, 9-12.09.2025)
CTTC IX - Current Trends in Theoretical Chemistry IX (Kraków, Poland, 1-5.09.2024)

ICESAA - International Conference on Excited State Aromaticity and Antiaromaticity
(Sigtuna, Sweden, 30.07-02.08.2019)

Aromaticity 2018 (Riviera Maya, Mexico, 28.11-01.12.2018)
CTTC VI - Current Trends in Theoretical Chemistry VI (Kraków, Poland, 1-5.09.2013)

ABOUT US PROJECTS PUBLICATIONS SOFTWARE AROMATICITY